







Hypertrophic cardiomyopathy (HCM) is a hereditary primary myocardial disease that is most commonly caused by mutations within genes encoding sarcomeric contractile proteins and is characterised by left ventricular hypertrophy in the absence of a cardiac or systemic cause.1,2 The condition is inherited as an autosomal dominant trait and has a prevalence of one in 500.3,4 Marked genetic heterogeneity, diverse clinical phenotypes and a highly variable natural history are well recognised.4–6 Although the overall prognosis is relatively good with an annual mortality rate <1 %, the propensity to potentially fatal ventricular arrhythmias is the most feared complication particularly because the peak incidence of sudden cardiac death (SCD) is during adolescence and early adulthood.6–9 The association with SCD is most frequently highlighted when a young, previously asymptomatic athlete falls victim and HCM is considered the commonest cause of SCD in young athletes worldwide.10–12
The arrhythmogenic substrate comprises left ventricular hypertrophy, myocyte disarray and interstitial fibrosis.13–17 Triggers for arrhythmias may include myocardial ischaemia, excessive sympathetic stimulation, left ventricular outflow tract obstruction (LVOTO) and paroxysmal atrial fibrillation (AF).18–20 The identification of patients at risk of arrhythmogenic SCD is an essential component in disease management given that the implantable cardioverter defibrillator (ICD) is the most effective therapy in preventing SCD.21–25 However, the low risk of adverse events in most patients coupled with the complex and unpredictable relationship between the arrhythmic substrate and triggers for arrhythmias means that risk stratification for arrhythmogenic SCD is a challenging aspect of the disease. A low threshold to implant an ICD into most patients with HCM is not cost-effective and is hampered by the high prevalence of inappropriate shocks and other complications relating to the implantation of the ICD.23–26
Aborted SCD and malignant ventricular arrhythmias are the most powerful risk factors for SCD.27–30 Patients who survive an episode of ventricular tachycardia (VT) or ventricular fibrillation (VF) remain at high risk of recurrent arrhythmogenic events, having an estimated risk of 10.6 % per annum and both the American College of Cardiology Foundation/ American Heart Association (ACCF/AHA) and American College of Cardiology/European Society of Cardiology (ACC/ESC) management guidelines for HCM recommend ICD implantation in such patients.27–30
Conventional Risk Factors for SCD in HCM
The selection of patients who may benefit from ICD therapy for primary prevention purposes is more challenging. Several potential risk factors for SCD have been reported, however conventionally regarded major risk factors include unexplained syncope, family history of sudden cardiac death (FHSCD), severe left ventricular hypertrophy (LVH), non-sustained VT (NSVT) on the Holter-monitoring or during exercise and an abnormal blood pressure response to exercise (ABPRE) (see Table 1).19,28–32 Consistent with the clinical diversity of the disease, all of these risk factors have a relatively low positive predictive accuracy in the range of 20 %.29 Conversely, each factor has an excellent negative predictive accuracy therefore a patient who does not exhibit any of these risk factors is suitably deemed low risk.29,31 Nevertheless, up to 3 % of arrhythmogenic SCDs occur in patients who do not exhibit any of these risk factors.33,34
The significance of these risk factors is governed by age.32–39 In young patients, syncope, severe LVH and NSVT are particularly associated with an increased risk of SCD.30,35–39 In older patients who have survived more than 60 years, the risk of arrhythmogenic SCD is low despite the presence of the five conventional risk factors above.39
Invasive electrophysiological studies, such as programmed ventricular stimulation, have a poorer predictive accuracy than some of the risk factors mentioned above and is not indicated for risk stratification.30 Paced ventricular electrogram fractionation analysis has been reported to reveal a positive predictive accuracy in the range of 38 %.40 However, the invasive nature of the procedure in combination with the dynamic nature of the risk profile of SCD in HCM patients means that periodical assessment is impractical.
Family History of SCD
A FHSCD from HCM in first-degree relatives of an affected patient or the presence of one or more premature SCD in the family has always been considered to represent an important risk factor because it is recognised that SCD events often cluster in families.29 Patients receiving an ICD for primary prevention based on a family history of HCM-related SCD experience appropriate electrical discharges comparable to other patient subsets with high-risk markers.41
Unexplained Syncope
Determination of the possible cause of unwitnessed syncope is challenging in HCM because there are multiple potential causes that include vasovagal syncope, arrhythmogenic syncope, abnormal vascular responses or transient severe mechanical LVOTO.28 However, the clinical perception is that syncope is the only premonitory cardiac symptom that is associated with SCD.28,42,43 Patients with syncopal events that occur in close temporal proximity (6 months) to the initial evaluation, show a substantially higher risk of SCD than patients without syncope.38 Older patients with remote syncopal events do not show an increased risk.38
Severe Left Ventricular Hypertrophy
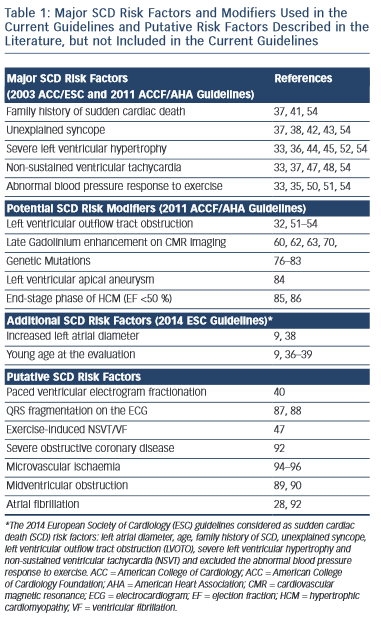
The severity and extent of LVH is associated with increased risk of SCD.30 Several studies have shown that a maximum wall thickness of ≥30 mm is associated with greatest risk of SCD.44–46 The ACCF/AHA guidelines state that the presence of extreme LVH alone is reasonable to recommend ICD29 (see Figure 1); however, extreme LVH is relative rare and the maximum wall thickness of a single segment may not adequately represent the true burden of hypertrophy.19 According to the 2003 ESC guidelines, the degree of maximum left ventricle (LV) wall thickness should be considered in the context of a multifactorial approach to risk stratification, rather than as an isolated risk factor.28,36 An exception may be the development of severe LVH at a young age (<18 years).36
Non-sustained Ventricular Tachycardia
The presence of repetitive ventricular arrhythmias, at rest or effort- induced is frequently used as a marker of increased electrical instability of the myocardium in clinical practice.37,47 NSVT (defined as ≥3 consecutive beats with a heart rate of ≥120 bpm) is detected in approximately 20 % of HCM patients and is associated with a substantial increase in SCD risk in young patients aged ≤30 years old.37 A relationship between the frequency, duration and rate of NSVT episodes has not yet been clearly demonstrated.37 In clinical practice isolated brief runs of NSVT on random Holter-monitoring rarely trigger decisions for prophylactic ICD, whereas frequent and/or prolonged (>10 beats) bursts of NSVT identified over serial monitoring periods, intuitively carry greater weight as a risk factor.48 In one study of 104 HCM patients with an ICD the presence of NSVT was the most predictive risk factor for appropriate ICD discharge in the 78 patients of the primary prevention group.24
Abnormal Blood Pressure Response to Exercise
Approximately one-third of patients with HCM, have an ABPRE (defined as either the failure to increase by at least 20 mmHg or a drop of at least 20 mmHg during effort), which can be due to central and peripheral mechanisms.28,29,49 An ABPRE rarely represents the sole indication for a prophylactic ICD implant in clinical practice and is usually considered in association with other risk factors (see Figure 1).29,35 The 2014 ESC guidelines did not consider the ABPRE as a risk factor since it has not been independently associated with SCD in any multivariate survival analysis (see Figure 2).29,30,50,51
Potential SCD Modifiers in HCM according to 2011 ACCF/AHA guidelines
Left Ventricular Outflow Tract Obstruction at Rest
Dynamic LVOTO is reported in approximately 25 % of patients during resting conditions.52,53 A study on 917 patients, including almost one-third with LVOTO, demonstrated an association between LVOTO and increased risk of SCD and appropriate ICD discharges over a 61-month median follow-up period.54 The risk of SCD was related to the severity of LVOTO and the presence of other recognised risk factors for SCD. Multivariable analysis demonstrated that LVOTO was an independent predictor of SCD/ICD discharge, with a 2.4-fold increase in the risk of SCD/ICD discharge.54 The role of provocable LVOTO with exercise is unclear and current guidelines do not recommend exercise-induced LVOTO in the risk stratification.30,55
Late Gadolinium Enhancement on CMR Study
The past decade has witnessed a burgeoning in the number of articles relating to the role of cardiovascular magnetic resonance (CMR) in HCM.56–76 In one study of 265 patients the quantification of LV mass correlated weakly with maximal wall thickness and was 100 % sensitive in predicting HCM-related mortality, but had a specificity of just 39 %.56 However, most of the interest in CMR is focused on the late enhancement after Gadolinium.57–72 Late Gadolinium enhancement (LGE) probably constitutes areas of myocardial replacement fibrosis and is detected in up to 60–70 % of HCM patients.57–63 As with all other aspects of the disease there is considerable heterogeneity in the extent and pattern of LGE.57–63 Fibrotic remodelling occurs early in disease pathogenesis of HCM but it may also be a secondary phenomenon related to microvascular ischaemia.13–17,64–66 Fibrous tissue represents a principal substrate for re-entrant ventricular arrhythmias and contributes to increased ventricular stiffness.59 A few studies have reported that the presence of LGE is significantly associated with heart failure death and all-cause mortality and is an independent predictor of adverse outcome and disease progression.59–62 In one study of 217 HCM patients the presence of fibrosis was associated with a 3.4-fold risk of major adverse events and the risk was proportional to the extent of LGE.59 Another study of 177 HCM patients showed that the presence of LGE may identify patients with increased susceptibility to ventricular tachyarrhythmias on the ambulatory Holter-monitoring (including a sevenfold increase in the risk of NSVT) and even small areas of LGE may be sufficient to promote arrhythmias.68 The significance of LGE in predicting arrhythmogenic SCD remains controversial. A recent metanalysis of four studies evaluating 1,053 patients, over an average follow-up of 3.1 years, concluded that LGE shows a trend towards significance for predicting SCD, but failed to shown a significant independent association.60 The high prevalence of LGE in HCM patients means that it would be impractical to consider it as a risk factor for SCD in isolation although extensive LGE has been shown to be associated with progressive ventricular dilatation and heart failure.58,59,69 Recently, two studies have provided conflicting results regarding the value of the extensive LGE in risk stratification for SCD.63,70 One study included 711 HCM patients, with a median follow-up of 3.5 years and 66 % of patients had LGE.63 The extent of LGE was found to be a strong univariable predictor of SCD, which was not maintained after adjustment for LV ejection fraction.63 The other study included 1,293 HCM patients, with a median follow-up of 3.3 years and presence of LGE in 42 % of patients. SCD events occurred in 37 patients (3 %), including 17 (1.3 %) with appropriate ICD discharge, which was considered equivalent to SCD.70 The extent of LGE was associated with an increased risk of SCD events and in particular LGE ≥15 % of the LV mass demonstrated a twofold increase in SCD event rate in those patients who would otherwise considered be at low risk. The authors concluded that extensive LGE provided additional information for assessing SCD event risk, particularly in HCM patients otherwise judged to be at low risk.70 The major criticism of this study was that even if the statistical analysis appeared to support this statement, the raw data did not.71 In the 20 patients that died suddenly or experienced aborted SCD only one revealed extensive LGE, while in the 17 patients experiencing an appropriate ICD shock, 13 patients had recognised conventional risk factors and from the rest only three had extensive LGE.70 In conclusion, the extent of LGE on CMR has some utility in predicting cardiovascular mortality, but the current data are contradictory and not conclusive in order to support the use of LGE in predicting the risk of arrhythmogenic SCD.30,71 Newer CMR techniques (as T1-mapping) may improve the characterisation of myocardial substrate of the arrhythmias.72–75
Additional Role of Genetics
Most sarcomere mutations capable of causing HCM are novel and limited to individual families; therefore, genetic screening is of limited value in risk stratification in most cases.4,5,29,30,76,77 The presence of multiple mutations or specific mutations encoding troponin T and lysosomal- associated membrane protein-2 (LAMP-2) may be indicative of a high risk of fatal events.78–83
Specific Cases
Left Ventricular Aneurysm
Left ventricular apical aneurysm with regional scarring is considered as a potential risk factor for primary prevention and has recently been reported in a high-risk subset of HCM patients.84 Left ventricular apical aneurysm is rare (approximately in 2 % of HCM patients) and is best characterised by CMR imaging. In one study of 28 patients, almost half of the patients with left ventricular apical aneurysm experienced adverse disease complications (event rate 10.5 %/year), including SCD, appropriate ICD discharges, non-fatal thromboembolic stroke and progressive heart failure and death.84
End-stage Phase of HCM
End-stage phase of HCM affects 3–8% of individuals and is characterised by progressive thinning of the myocardium with cavity enlargement and impaired systolic function.85,86 The complication is a result of extensive and transmural fibrosis and has a high incidence of SCD with an annual mortality rate exceeding 10 %.85,86 In such patients, prophylactic ICD implantation is a generally accepted clinical practice.29,85,86
2003 ACC/ESC Guidelines versus 2011 ACCF/AHA Guidelines
The main disagreement between the US and Europe guidleines is historically based on the number of risk factors required before consideration of an implantation of an ICD for primary prevention.28,29 Given the low positive predictive value of each of the conventional risk factors, the European approach has been to implant an ICD only in the presence of >1 risk factor.28 By contrast, the US approach recommends ICD implantation patients with FHSCD from HCM in a first-degree relative, LV wall thickness ≥30 mm or recent unexplained syncope as isolated risk factors whereas those patients with NSVT or an ABPRE require another risk factor or risk modifier (such as LVOTO, LGE on CMR imaging, LV apical aneurysm or a high-risk genetic mutation)29 (see Figure 1). This difference in approach is partially due to the conflicting results between American and European studies regarding the risk stratification.22,33 Previous American studies have reported that an important proportion of discharges occur in patients implanted with a prophylactic ICD with just one risk factor.22 The European concern is that if ICDs were inserted in all patients with one risk factor the incidence of device complications would surpass the potential benefits.33,34 Although there is no doubt about the value of an ICD in preventing SCD with appropriate discharge rates ranging from 2–3.6 %/year for primary prevention cases and 4.3–10.6 %/ year for secondary prevention cases, the inappropriate shock rate and implant complications range from 16–27 % and 12–18 %, respectively.22,23 A recent meta-analysis of 16 HCM cohorts reported inappropriate ICD interventions and complication of 4.8 %/year and 3.4 %/year, respectively.25
HCM Risk-SCD Model of the 2014 ESC Guidelines
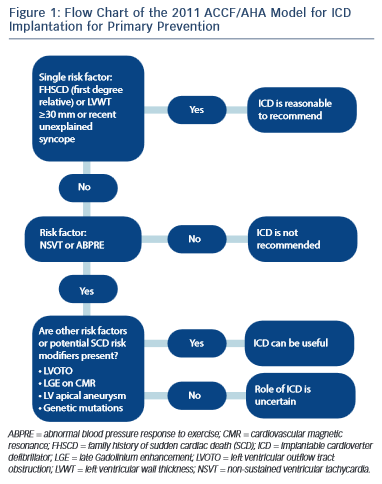
A fundamental problem with the aforementioned risk stratification procedure is the assumption that the significance of all of the risk factors remains static throughout life. Furthermore, important parameters such as LVH or LVOTO, which are contiguous variables, are treated as binary factors (present or absent). The ESC HCM outcome investigators have recently recommended a 5-year risk calculator derived from a model involving a large retrospective longitudinal multicentre experience from 3,675 patients.9,30 Eight clinical parameters were included as pre- specified predictors that were independently associated with SCD in at least one published study of multivariate survival analysis. Of the eight parameters, seven were associated with SCD or an appropriate ICD shock at the 15 % significance level and these were: age, FHSCD, maximal LV wall thickness, left atrial diameter, maximal LVOTO, NSVT and unexplained syncope (see Figure 2).
The incorporation of these parameters into the model equation is used to estimate the 5-year risk of SCD for any particular patient. The cut-off level of ≥6 % SCD risk in 5 years is recommended for considering an ICD implant for primary prevention. Individuals with a risk score of <4 % are considered as low risk, whereas those with a risk score of 4–6 % of SCD characterise an intermediate-risk group where ICD may be considered (see Figure 2).
The results of this study indicate that the use of this model is superior to prior conventional methods for detecting high-risk individuals previously considered at low or intermediate risk. The model also scores highly for correctly identifying individuals at high risk of SCD.30
The new 2014 ESC risk stratification model is limited to some extent in that it was not validated in paediatric patients (<16 years), in patients with syndromic LVH or in a large population of non-Caucasian individuals. Furthermore the effect of latent LVOTO or the effect of LVOTO reduction by alcohol ablation or myectomy was not tested and very few patients had extreme LVH ≥35 mm.30
Other Potential Risk Factors and Arbitrators not Included in the Current Guidelines
A number of electrical, structural and functional markers that can be assessed using simple investigations have been proposed for risk stratification. The fragmentation of the QRS on the ECG has been postulated to predict ventricular arrhythmic events.87,88 A study in 167 patients with a mean follow-up of 6.3 years, fragmentation of the QRS was a strong independent predictor for major arrhythmic events including SCD.87
There are reports that HCM associated with midventricular obstruction (with pressure gradient ≥30 mmHg) may be an independent predictor of adverse outcomes, especially the combined endpoint of SCD and potentially lethal arrhythmic events.89,90 Conversely, apical HCM has been associated with a benign prognosis.91
Myocardial ischaemia that may be caused by small vessel disease or concomitant severe epicardial coronary artery disease has also been considered as possible risk factor.28,92,93 In one study of 433 HCM patients, 27 % had severe epicardial CAD and this was a significant predictor for cardiac death and SCD.92 Assessment of coronary microvascular dysfunction is challenging and stress perfusion CMR imaging could have a future role in risk stratification.94–96 Exercise capacity has also been proposed to help risk stratification in different studies.97–99 It has been demonstrated that peak VO2 is associated with an increased risk of major events during short-term follow-up.99
In conclusion, there have been significant advances in the risk stratification of HCM since the disease was first described over 5 decades ago. The heterogeneous nature of the disease and the variation in trigger factors provides an adequate explanation for the low predictive accuracy of most conventional risk factors in isolation. A new risk model for risk stratification proposed by the ESC HCM outcome group shows promise but requires validation in different cohorts. The ICD is the only effective therapy in preventing SCD for the disease with a relatively low adverse event rate, but most deaths occur in relatively young patients. However, it is also difficult to ignore the complications with the ICD, therefore, the strife to perfect risk stratification in HCM should continue to ensure that only the most high-risk patients receive an ICD.